Interactions pharmacocinétiques
Une interaction médicamenteuse pharmacocinétique (IMPK) résulte de l’action d’un médicament qui va modifier les caractéristiques pharmacocinétiques d’un autre médicament.
Les IMPK se produisent aux points clés du devenir d’un médicament dans l’organisme que sont l’absorption intestinale, la diffusion, le métabolisme hépatique et l’excrétion biliaire ou rénale comme indiqué dans le tableau 1 et la figure 1.
Figure 1 : les sites d'action des interactions médicamenteuses
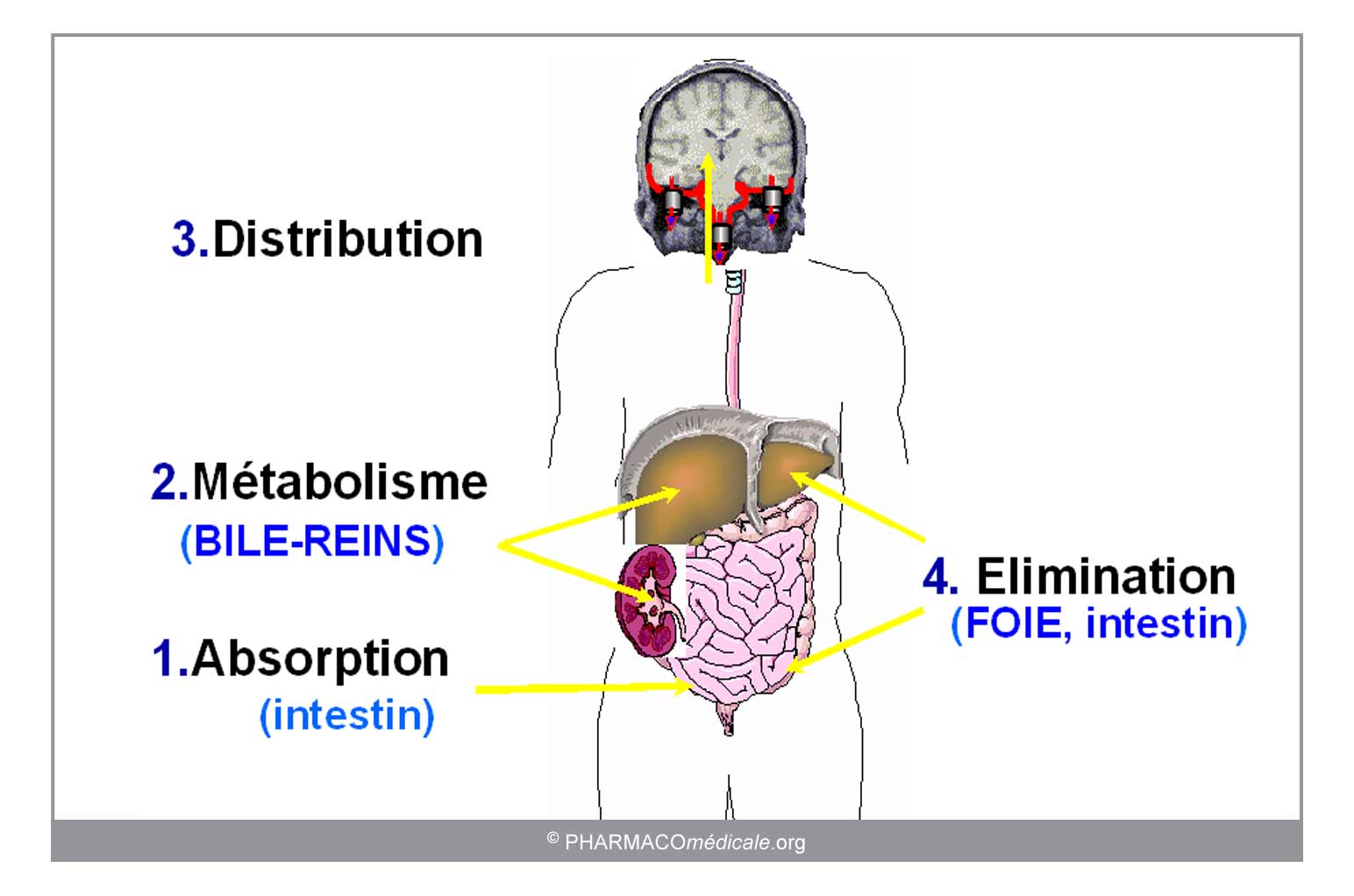
Les principaux mécanismes impliqués font appel à l’inhibition soit d’enzymes du métabolisme des médicaments, soit à des protéines de transport de médicaments ou à l’induction d’enzymes ou de transporteurs.
D’autres mécanismes sont aussi impliqués dans l’intestin : l’augmentation du temps de contact du médicament dans l’intestin (ralentisseurs du transit intestinal ou anti émétiques) augmente l’absorption des médicaments associés; des élévations de pH gastroduodénal (anti ulcéreux) diminuent la fraction non ionisée des acides faibles, essentiellement absorbés dans l’estomac et le duodénum et donc en limitent l’absorption. Certains pansements gastriques représentent une véritable barrière physique tapissant toute la surface de l’intestin et empêchent l’absorption des médicaments.
| Mécanisme(s) | Exemples de médicaments responsables | Conséquences | |
| Absorption intestinale |
Ralentisseurs du transit intestinal |
Anti-émétiques ou antidiarrhéiques (métoclopramide lopéramide) |
Augmentation de F, AUC, Cmax, demi-vie inchangée. Toxicité accrue |
| Elévation du pH intestinal et diminution de la fraction non ionisée des acides faibles | oméprazole |
Diminution de F, AUC, Cmax, demi-vie inchangée Inefficacité |
|
| Barrière physique gastro-duodénale | Pansements gastriques |
Diminution de F, AUC, Cmax, demi-vie inchangée Inefficacité |
|
| Inhibition d’un transporteur d’efflux (lien cours G17-5) | Inhibiteurs de P-glycoprotéine (quinidine, ritonavir) |
Augmentation de F, AUC, Cmax, demi-vie inchangée Toxicité accrue |
|
| Inhibition du métabolisme intestinal des médicaments | Inhibiteurs CYP3A4 (clarythromycine) |
Augmentation de F, AUC, Cmax, demi-vie inchangée Toxicité accrue |
|
| Diffusion | Inhibition (ou induction) de transporteurs (BHE) | Inhibiteurs de P-glycoprotéine (quinidine, ritonavir) |
Augmentation des concentrations dans un organe (cerveau) Toxicité accrue Efficacité accrue (anticancéreux, antirétroviraux) |
| Métabolisme | Inhibition (lien cours G17-2) ou induction (lien cours G17-3) du métabolisme hépatique | Inhibiteurs (lien cours G17-2) ou inducteurs (lien cours G17-3) de CYP ou autres enzymes hépatiques |
Diminution (inducteurs) ou augmentation (inhibiteurs) AUC, Cmax et demi-vie Toxicité accrue |
| Excrétion | Inhibition de sécrétion rénale ou biliaire | Inhibiteurs de transporteurs d’efflux ou d’influx tubulaires rénaux ou hépatocytaires |
Augmentation AUC, Cmax et demi-vie Toxicité accrue |
Tableau 1 : Interactions médicamenteuses pharmacocinétiques
F : biodisponibilité orale ; AUC : Aire sous la courbe des concentration plasmatiques d’un médicament en fonction du temps ; Cmax : concentration plasmatique maximale ; CYP3A4 : cytochrome P450 3A4 ; BHE : barrière hémato encéphalique
Conséquences des IM pharmacocinétiques :
Les IMPK qui modifient l’absorption intestinale des médicaments modifient l’aire sous la courbe du médicament en fonction du temps (AUC) et/ou la concentration maximale (Cmax). Si les processus de métabolisme hépatique et d’excrétion biliaire et rénale ne sont pas modifiés, la demi-vie reste inchangée.
Les IMPK qui résultent d’une inhibition du métabolisme hépatique et/ou de la sécrétion rénale ou biliaire augmentent l’AUC, la Cmax et la demi-vie.
Les IMPK qui résultent d’une induction du métabolisme hépatique et/ou de la sécrétion rénale ou biliaire diminuent l’AUC, la Cmax et la demi-vie.
Les IMPK sont à risque de conséquence clinique lorsqu’elles affectent un médicament à index thérapeutique étroit, c’est à dire dont les seuils de toxicité par surdosage et d’inefficacité par sous dosage sont proches (ex anticancéreux, immunosuppresseur, anti arythmiques...) comme illustré dans la figure ci-dessous.
Figure 2.

Pour les médicaments dont l’index thérapeutique est large, les conséquences cliniques de modifications de concentrations plasmatiques sont en général absentes et ne sont pas signalées ou bénéficient d’un niveau de précaution faible.
En général les surdosages exposent à une fréquence accrue d’effets indésirables et les sous dosages à une inefficacité thérapeutique. Cependant, les conséquences cliniques des IM affectant les enzymes ou les transporteurs dépendent des propriétés pharmacodynamiques propres du médicament inchangé et de ses métabolites. Par exemple, une inhibition du CYP2D6 s’accompagnera d’une diminution de l’effet antalgique de la codéïne puisque celle-ci ne sera pas métabolisée en morphine, métabolite actif sur lequel repose l’effet attendu. Ainsi, pour un même phénomène (ex. inhibition d’une enzyme), les conséquences pharmacologiques peuvent être opposées selon le médicament concerné (augmentation ou diminution de l’effet pharmacologique)
Quand et pendant combien de temps les IMPK se manifestent-elles ?
Les interactions résultant d’un effet direct sur les fonctions gastrointestinales se manifesteront immédiatement et dureront tant que l’altération de transit existera.
Les interactions par inhibition d’une enzyme ou d’un transporteur sont elles-aussi immédiates. En effet, le plus souvent, l’inhibition résulte d’un mécanisme compétitif ; l’interaction apparaitra dès que les deux substances seront en présence et disparaitra lorsque le médicament interférent aura atteint des concentrations sub-inhibitrices. La durée de l’interaction dépend donc de la demi-vie du médicament inhibiteur
Les interactions par induction d’une enzyme ou d’un transporteur apparaissent avec un délai correspondant au temps nécessaire à la synthèse de la protéine puisque le mécanisme de l’induction enzymatique est transcriptionnel. Ainsi, l’effet inducteur n’apparaitra qu’au bout de quelques jours et durera également quelques jours après l’arrçet de l’inducteur (turn-over de la protéine enzymatique).
Cas particulier : Mise à profit des IMPK pour améliorer les caractéristiques pharmacocinétiques de médicaments :
Les processus de métabolisme et de transport des médicaments sont à l’origine d’une grande variabilité de la pharmacocinétique d’un individu à l’autre. De plus, le métabolisme présystémique peut parfois considérablement limiter la biodisponibilité orale (F) de certains médicaments, obligeant à donner de très fortes doses plusieurs fois dans la journée.
- Les antirétroviraux de la classe des inhibiteurs de la protéase (antiprotéase) ont pour la plupart des biodisponibilités orales (F) très faibles du fait d’un important métabolisme présystémique. Pour éviter des les administrer plusieurs fois par jour à des posologies très importantes, les entreprises du médicament les administrent maintenant systématiquement avec un très puissant inhibiteur de cytochromes P450 hépatiques, le ritonavir. Le ritonavir est également un antirétroviral dirigé contre le HIV, mais il est administré à de faibles doses uniquement pour inhiber des CYP et pas à visée antirétrovirale. L’adjonction systématique de ritonavir avec les antiprotéase a permis d’une part d’augmenter considérablement leur biodisponibilité orale (multipliée par 36 pour le saquinavir qui, utilisé seul a une F de 4 %) mais également d’atteindre des concentrations plasmatiques plus efficaces. La diminution du nombre de prises permet également une meilleure observance.
Figure 3 : interactions médicamenteuses saquinavir/ritonavir

Le probénécide a été développé pour limiter l’élimination urinaire de la pénicilline à une époque où la production de pénicilline par synthèse n’était pas suffisante. Le probénécide bloque la sécrétion tubulaire rénale de médicaments et permet ainsi de diminuer leur clairance rénale. Le probénécide est encore utilisé de nos jours pour prévenir la néphrotoxicité d’un antiviral (agissant sur le CMV), le cidofovir. Le cidofovir est activement sécrété par les tubules rénaux et est donc retrouvé à de fortes concentrations tissulaires intra rénales, toxiques pour les cellules tubulaires. Afin de prévenir l’accumulation de cidofovir dans les reins, on l’associe toujours à du probénécide qui permet l’utilisation de cet antiviral sans toxicité rénale importante. Il a été proposé qu’en cas d’épidémie de grippe aviaire, le seul antiviral disponible à l’heure actuelle, (oséltamivir) pourrait être associé au probénécide afin de pouvoir diminuer sa posologie et le distribuer au plus grand nombre de patients.
- Dernière mise à jour le .