ESSAIS CLINIQUES CHEZ L'HOMME | PHASE I | PHASE II | PHASE III
Essais précoces (Phases I et II)
Les essais précoces constituent la première étape du développement clinique chez l’Homme. Ils permettent d’évaluer successivement : la tolérance et la sécurité d’emploi (phase I) et la première démonstration d’efficacité (phase II).
Les essais de phase I ne peuvent être réalisés que dans des centres autorisés, disposant d’une habilitation spécifique, d’équipements adaptés et d’une surveillance médicale renforcée afin d’assurer la sécurité des participants.
Les participants à des essais de phase I peuvent recevoir une indemnisation financière destinée à compenser les contraintes liées à leur participation (temps consacré, déplacements, examens, hospitalisation éventuelle). Cette indemnisation ne constitue pas une rémunération au sens d’un travail salarié et son montant est réglementé afin d’éviter toute incitation excessive.
Les volontaires sains ne peuvent pas participer à plusieurs essais successifs sans respecter une période d’exclusion (« wash-out ») afin de limiter les risques liés aux expositions répétées.
|
Phase |
Participants à la recherche |
Dose |
Mode d'administration |
Nombre |
Type |
Objectifs |
|
I |
Sujets sains (sauf si molécule trop toxique pour être administré à une personne saine, ex : anticancéreux) |
Escalade de doses / dose unique et répétée |
Plusieurs formes galéniques pour comparer plusieurs voies d'administration |
Petit effectif (10 - 100 personnes) |
- Sans bénéfice individuel direct - Durée courte |
- Dose maximale tolérée (DMT) - Sécurité d'emploi - Propriétés identiques à celles observées chez l’animal ? - Mode d'administration (voie, intervalle, forme galénique, influence des repas) - Pharmacocinétique chez l'Homme sain (prévisions des précautions à prendre et des populations à étudier, métabolites...) |
|
II |
Sujets sains (IIa) ou malades (IIb), groupes homogènes et sélectionnés |
Plusieurs doses |
Recherche de la forme galénique définitive |
Petit effectif (10 - 100 personnes) |
- Avec bénéfice individuel direct pour les malades - Durée courte - Pas de groupe de comparaison |
- Sécurité d'emploi à court terme chez le patient - Dose minimale efficace (DME) - Relation dose-effet et concentration-effet - Effet de la maladie sur le médicament - Pharmacocinétique - Recherche du schéma posologique utilisé en phase III (ou bien dans une éventuelle étude IIb) |
Dans certaines situations particulières, notamment lorsqu’il existe un besoin thérapeutique non couvert pour des maladies graves (exemple : nouvelle classe pharmaco-chimique pour des maladies graves comme les cancers, etc …), des médicaments non encore commercialisés peuvent être mis à disposition avant l’obtention de leur AMM, après réalisation des études pilotes et après autorisation des autorités sanitaires (en France, l’ANSM). Pour cela, deux mécanismes existent :
- Accès précoce : destiné à des médicaments présumés innovants, pour lesquels l’efficacité et la sécurité sont déjà suffisamment établies mais dont l’AMM ou le remboursement n’est pas encore disponible.
- Accès compassionnel : destiné à des patients sans alternative thérapeutique adaptée, pour des médicaments ne bénéficiant pas encore d’une indication autorisée ou dont les données sont plus limitées.
Si les résultats des phases I et II sont favorables, le candidat médicament passe en phase III afin de confirmer son efficacité et sa sécurité à grande échelle avant la demande d’AMM.
Essais de phase III
Les essais de phase III constituent des études confirmatoires réalisées avant la demande d’AMM. Ils visent à confirmer l’efficacité thérapeutique observée en phase II, à confirmer la sécurité d’emploi, à préciser le rapport bénéfice/risque, à définir la place du médicament dans la stratégie thérapeutique.
|
Phase |
Participants à la recherche |
Dose |
Mode d'administration |
Nombre |
Type |
Objectifs |
|
III |
Malades |
Posologie que l'on souhaite commercialiser, définie à partie des essais de phase II (IIb) |
Forme galénique définitive |
Grand nombre (plusieurs centaines), population cible du médicament |
Durée assez longue (plusieurs mois, voire années) |
- Efficacité et sécurité d'emploi - Détermination du rapport bénéfice/risque - Interactions potentielles - Comparaison au médicament de référence (placebo s’il n’existe pas de traitement de référence) - Obtention de l’indication - Extension d’indication (IIIb) |
Aspects méthodologiques
L’objectif de l’essai de phase III est d'apporter la preuve scientifique de l'efficacité clinique des traitements. Il permet la mise en place de thérapeutiques sur la base de faits avérés et non pas sur des hypothèses ou des raisonnements théoriques.
Un essai clinique doit respecter un protocole de recherche clinique. Il doit être rédigé au préalable, avant de débuter l’essai. Il définit la question, cliniquement pertinente, précise et clairement formulée, à laquelle il doit répondre, qui correspond à l’objectif de l’essai, ainsi que le plan expérimental (modalités pratiques de l’essai) et les techniques d’analyse des données qui seront utilisées.
1. Sélection des sujets
L’ensemble des sujets recrutés doit être représentatif de la population cible pour permettre l’extrapolation des résultats à l’ensemble de cette population. Il doit constituer une population homogène afin de réduire la variabilité dans la réponse au traitement.
Le recrutement des sujets doit se faire selon les critères d’inclusion et de non-inclusion définis au préalable et de manière non-ambiguë. Les critères d’inclusion sont des caractères que les sujets doivent obligatoirement présenter pour être inclus, et les critères de non-inclusion sont des caractères que les sujets ne doivent pas présenter pour être inclus. Il ne faut pas confondre les critères de non-inclusion avec les critères d’exclusion, qui sont des éléments, eux aussi définis au préalable, qui conduisent à retirer un patient d’une étude en cours.
2. Nombre de sujets à inclure
Le calcul de ce nombre tient compte de :
- l'objectif de l'essai (la plus petite différence que l'on souhaite détecter ce qui nécessite de définir le bénéfice attendu ; plus la différence recherchée entre un médicament et son comparateur est petite, plus il faut inclure de patients) ;
- la variabilité du critère de jugement dans la population ;
- la puissance de l'essai (probabilité de détecter une différence quand elle existe et qui donnera plus ou moins de crédibilité aux résultats futurs de l'essai, la puissance est égale à 1-β où le risque β, appelé risque de deuxième espèce est compris entre 80 et 90%);
- la part laissée au hasard de détecter une différence entre les deux groupes sans lien avec l'efficacité du traitement (risque α, souvent fixé à 5%, ou celui de détecter une différence qui n'existe pas).
Généralement, les essais sont multicentriques, c'est-à-dire que le protocole est réalisé dans plusieurs structures distinctes sur une même période de temps, la coordination étant assurée par un investigateur coordinateur (1 par pays participant). Ce type d'essai nécessite une standardisation des méthodes d'évaluation, du suivi de l'étude et bien évidemment du protocole, mais permet d’inclure plus facilement.
3. Plans expérimentaux
Seuls les essais comparatifs permettent de conclure à l’efficacité d’un traitement. Le médicament étudié est comparé soit :
- versus placebo (traitement dénué du principe actif et donc d’activité propre) : utilisé lorsqu’il n’existe pas de traitement de référence, permet d’estimer l’effet propre du médicament. Le but est alors de montrer la supériorité du médicament au placebo.
- versus un traitement de référence : utilisé lorsqu’un traitement efficace existe déjà, permet de déterminer si le nouveau médicament est supérieur, non inférieur, ou équivalent par rapport au médicament de référence.
L’évaluation est généralement réalisée entre deux groupes de sujets : un groupe recevant le traitement et un groupe recevant le placebo (ou le traitement de référence).
Cette comparaison doit se faire selon un schéma précis. Plus particulièrement, l'attribution des traitements doit se faire par tirage au sort afin de répartir les patients dans les différents groupes de traitement de façon aléatoire : c’est la randomisation. Cette randomisation permet d’assurer la comparabilité des groupes et de conclure que, s’il existe une différence entre les groupes, celle-ci n’est due qu’au traitement.
Dans la mesure du possible, afin d'éviter tout biais de jugement, l’étude se déroulera en double-aveugle (ou double-insu) : ni le patient, ni le médecin ne connaîtront le traitement attribué. Lorsque le double insu n’est pas possible, l’étude peut être réalisée en simple insu (le médecin connaît alors le traitement reçu par le patient) ou en ouvert lorsque patient et investigateur connaissent tous les deux les traitements administrés.
La comparaison entre deux groupes peut se faire en groupes parallèles (il s'agit alors d'un essai contrôlé où un des groupes joue le rôle de témoin) ou en cross-over (ou essai croisé). Un plan d’étude en cross-over augmente la puissance statistique de l’essai par rapport à un plan d’étude en groupes parallèles.
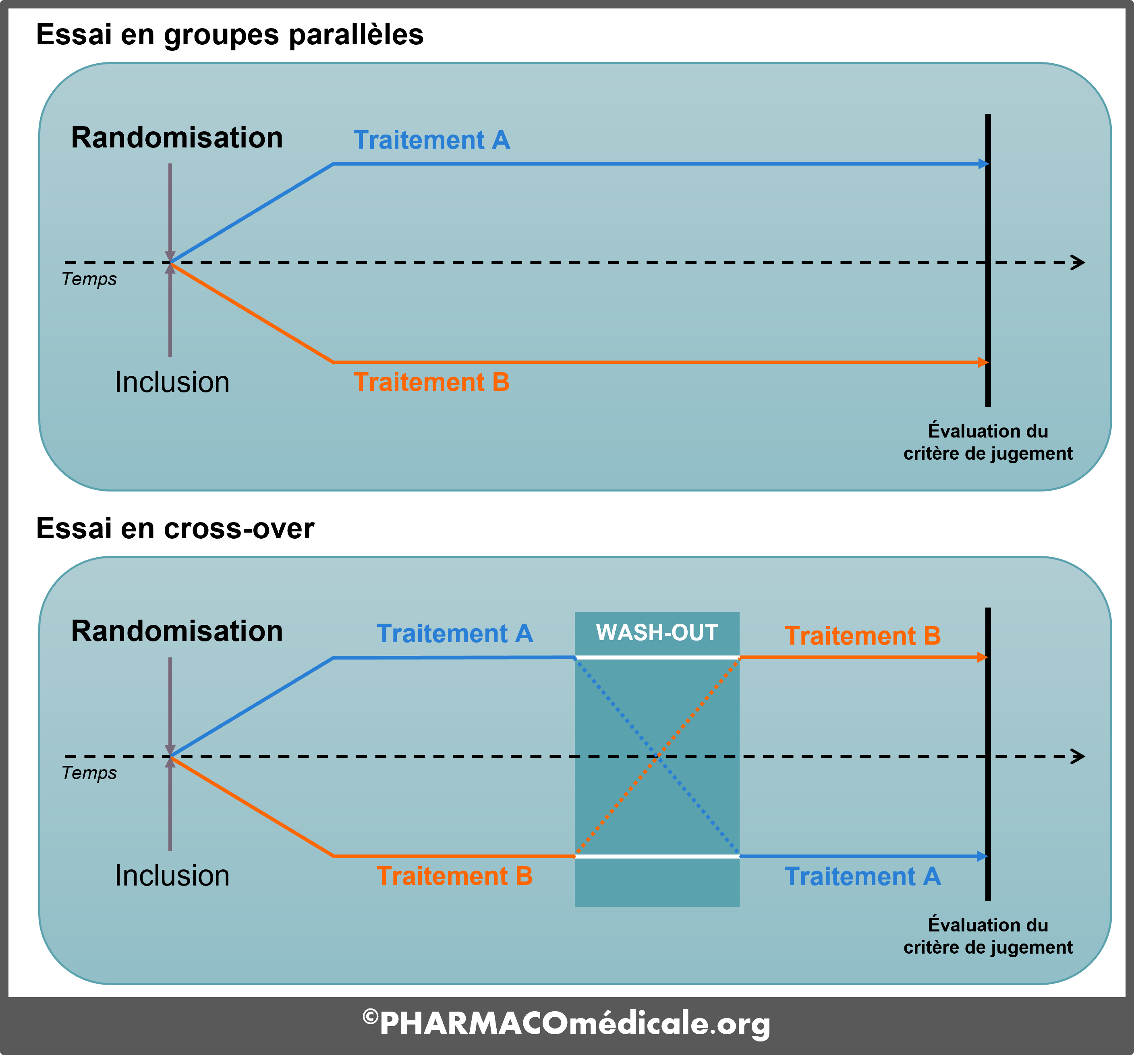
L'évaluation de l'efficacité de la molécule se fait grâce à un critère d'évaluation ou critère de jugement principal. Il s’agit d’un élément unique, cliniquement pertinent, objectif (mesurable et reproductible) et si possible direct. Un critère de substitution pourra être utilisé en cas d’évaluation directe inaccessible.
Le recueil des données se fait dans des cahiers d'observation (ou CRF pour « Case Report File »).
Au cours de l’étude, les investigateurs sont dans l’obligation de déclarer les événements indésirables graves au promoteur.
Au moment de l’exploitation des résultats de l’essai thérapeutique, toute déviation au protocole doit être signalée. Des patients peuvent être considérés comme « perdus de vue » dès lors qu'ils n'ont plus donné suite aux visites chez l'investigateur et que personne n'a eu de nouvelles. Il est très important de chercher à retrouver par tous les moyens ces personnes dont les résultats seront analysés mais qui, s'ils sont nombreux, entacheront toujours d'erreur le résultat global de l'étude.
4. Analyse des résultats
Les résultats de l'étude devront être analysés de façon très rigoureuse selon des tests statistiques adaptés, choisis au préalable. L’effet observé doit non seulement être statistiquement significatif mais il doit également être cliniquement pertinent.
L’ensemble de la population incluse dite en « intention de traiter » (ITT ou FAS pour Full Analysis Set) doit être analysé pour pouvoir conclure à un effet de la molécule testée. Toutefois, l'analyse « per protocole » (PP) des résultats qui tient compte des sorties d'étude, de l'observance du traitement et des éventuelles déviations au protocole peut être retenue. Parfois, des analyses de sous-groupes permettent de préciser l'effet du médicament (ces analyses doivent avoir été prévues a priori lors de la rédaction du protocole).
Avantages et limites des essais de phase III
Les essais randomisés contrôlés constituent la référence méthodologique (gold standard) pour l’évaluation des traitements.
Ils présentent les avantages suivants : réduction des biais de sélection, meilleure comparabilité des groupes, forte validité interne, démonstration robuste d'une relation causale entre traitement et effet observé, haut niveau de preuve scientifique.
Cependant, ils présentent également des limites : critères d’inclusion souvent très stricts, populations peu représentatives de la pratique réelle (personnes âgées, patients polypathologiques, femmes enceintes, enfants), durée parfois insuffisante, détection limitée des effets indésirables rares (<1/1000 ou 1/10 000 voire moins), coût élevé, contraintes logistiques importantes. La conséquence consiste en une efficacité démontrée en essai clinique ne reflète pas toujours exactement l’efficacité observée en pratique courante (effectiveness versus efficacy).
Pour aller plus loin
Pour approfondir les enjeux liés au développement et à l’évaluation des médicaments, le Livre Blanc élaboré par le groupe de travail « Méthodologie » de la Société Française de Pharmacologie et Thérapeutique (SFPT), intitulé « De la nécessité de la méthodologie dans l’évaluation des médicaments » est disponible gratuitement ici : https://sfpt-fr.org/livreblancmethodo/index.htm
Ce document propose une vue d’ensemble des principes méthodologiques qui justifient les conditions de réalisation des essais cliniques et contribue ainsi à développer un regard critique sur les modalités de mise à disposition de nouveaux médicaments. Bien qu’il aborde certains aspects techniques, il demeure accessible à un large public. Les lecteurs souhaitant approfondir certains points pourront également s’appuyer sur les documents compagnons associés. Ce Livre Blanc vise à rendre les concepts fondamentaux des essais cliniques plus accessibles et à rappeler que ces principes constituent des outils essentiels pour l’évaluation des médicaments comme des thérapeutiques non médicamenteuses.
- Dernière mise à jour le .