
Les points essentiels
1- Définitions et intérêts des paramètres pharmacocinétiques pour la prescription.
Le schéma posologique choisi pour un médicament a généralement pour but de maintenir les concentrations sanguines dans l’intervalle thérapeutique, c’est à dire dans la zone de concentrations assurant l’effet thérapeutique optimal tout en minimisant le risque d’effets indésirables (figure 1).
Figure 1: Intervalle thérapeutique d’un médicament.
Après administration d’un médicament, l’évolution des concentrations sanguines dépend :
- des modalités d’administration (voie d’administration, forme galénique, posologie…)
- des paramètres pharmacocinétiques qui décrivent, sur un plan quantitatif, le devenir du médicament dans l’organisme
Les paramètres pharmacocinétiques sont déterminés lors du développement clinique des médicaments (études de phase I) dans des groupes homogènes de sujets. Ils doivent également être définis dans des groupes de sujets ayant des caractéristiques physio-pathologiques variables (sujets âgés, patients insuffisants rénaux,…) afin de quantifier l’impact de ces facteurs individuels sur les valeurs des paramètres et donc sur l'évolution des concentrations.
Les principaux paramètres pharmacocinétiques sont :
- La biodisponibilité (F) d’un médicament est définie comme la fraction de la dose administrée ou du principe actif libéré par la forme pharmaceutique qui parvient sous forme inchangée dans la circulation sanguine systémique et la vitesse à laquelle se réalise ce processus. Elle pourra être modifiée par des facteurs affectant l’absorption digestive ou l’effet de premier passage hépatique.
- Le volume de distribution (Vd) est un volume fictif théorique, qui représente le volume dans lequel devrait se distribuer le médicament pour être à l’équilibre à la même concentration que dans le plasma.
Il représente la capacité d’un médicament à diffuser dans l’organisme. - La clairance (Cl) représente le volume sanguin ou le volume plasmatique totalement épuré du médicament par unité de temps. Cela correspond à la capacité de l’organisme à éliminer le médicament. La clairance pourra être modifiée par toute cause affectant l’élimination rénale ou hépatique du médicament.
- La demi-vie d'élimination (t1/2) est définie comme le temps nécessaire à la diminution de 50% de la concentration plasmatique. C'est un indicateur de la durée de persistance du médicament dans l’organisme; elle pourra être affectée par des modifications de clairance ou de volume de distribution.
Toute modification des paramètres pharmacocinétiques sous l’influence de facteurs physiologiques, pathologiques ou environnementaux s’accompagnera de différences dans l’évolution des concentrations (figure 2).
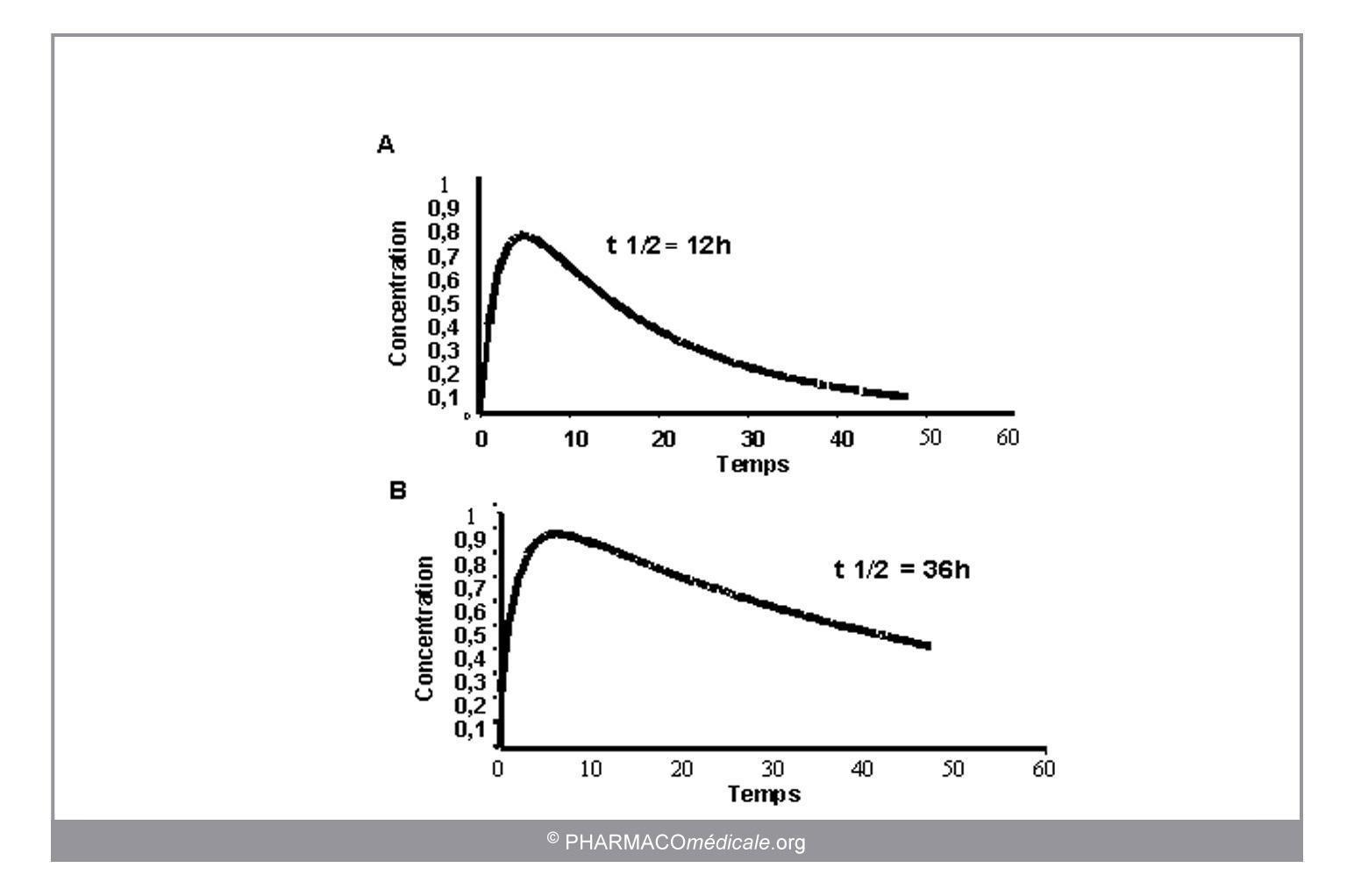 Figure 2 : Impact de la fonction rénale sur la demi-vie d’un médicament : évolution des concentrations suite à l’administration orale d’une même dose à un groupe de sujets normaux (A) et à un groupe de sujets insuffisants rénaux (B) avec une demi-vie 3 fois plus longue.
Figure 2 : Impact de la fonction rénale sur la demi-vie d’un médicament : évolution des concentrations suite à l’administration orale d’une même dose à un groupe de sujets normaux (A) et à un groupe de sujets insuffisants rénaux (B) avec une demi-vie 3 fois plus longue.
Lors de la prescription, l’adaptation individuelle de posologie repose sur la prise en compte des modifications attendues de la pharmacocinétique du médicament chez le patient traité (figure 3).
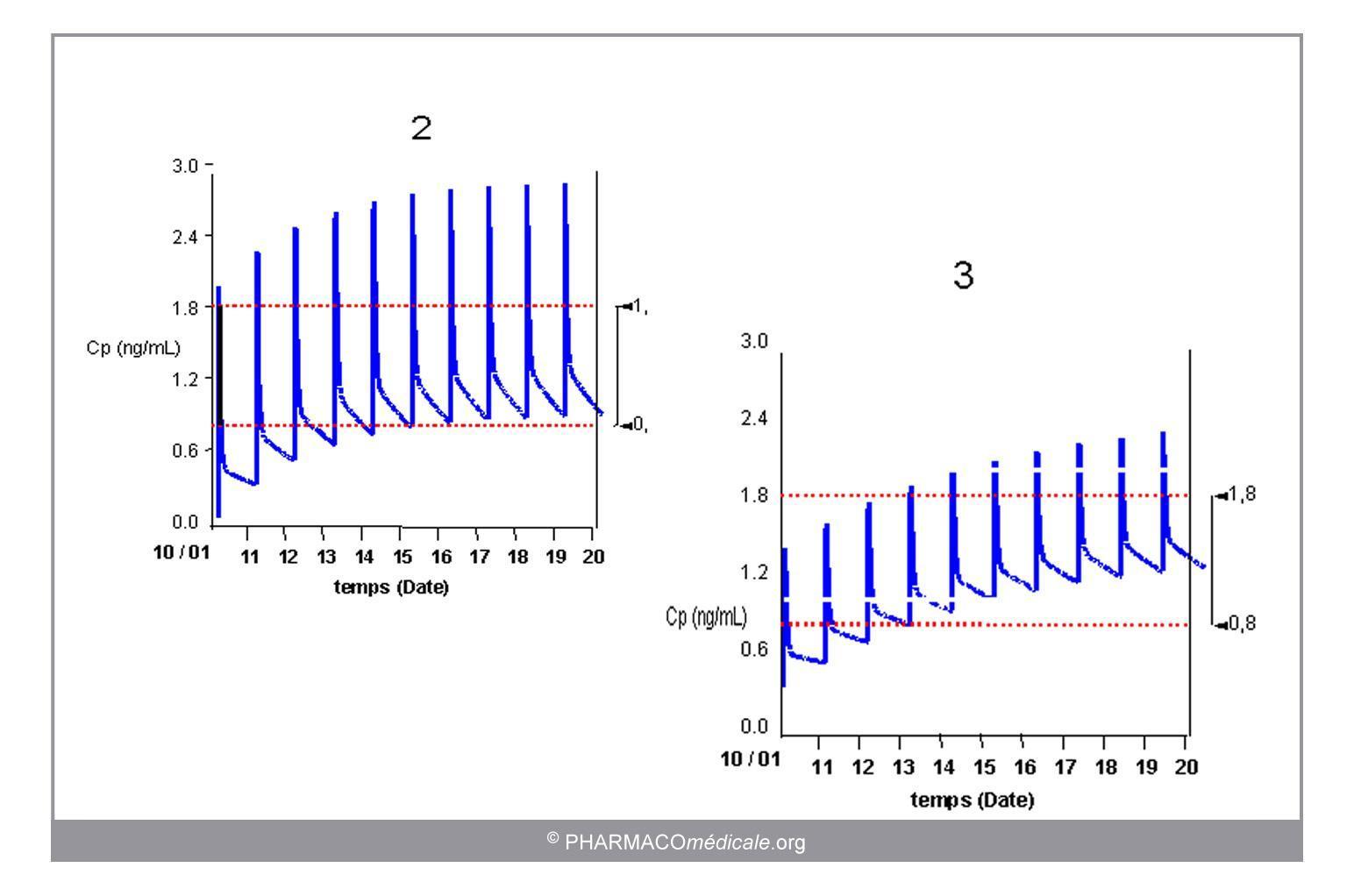 Figure 3 : évolution des concentrations sanguines d’un médicament prescrit à posologie standard et à posologie adaptée aux caractéristiques pharmacocinétiques individuelles. La posologie de digoxine permettant d’obtenir des concentrations résiduelles comparables est de 300µg/24h chez un sujet à fonction rénale normale (2) et de 125 µg/24h chez l'insuffisant rénal (3).
Figure 3 : évolution des concentrations sanguines d’un médicament prescrit à posologie standard et à posologie adaptée aux caractéristiques pharmacocinétiques individuelles. La posologie de digoxine permettant d’obtenir des concentrations résiduelles comparables est de 300µg/24h chez un sujet à fonction rénale normale (2) et de 125 µg/24h chez l'insuffisant rénal (3).
2- Principe du calcul des paramètres pharmacocinétiques.
Le calcul des paramètres pharmacocinétiques s’effectue à partir de données expérimentales obtenues chez l’homme suite à l’administration d’un médicament. Il s’agit de l'étude de l’évolution des concentrations sanguines du médicament au cours du temps.
Les paramètres pharmacocinétiques peuvent être calculés selon une approche compartimentale ou non compartimentale :
- Avec l’approche non-compartimentale, les paramètres pharmacocinétiques sont directement déduits des points expérimentaux (Cmax, Tmax) ou obtenus par l’intermédiaire d'équations mathématiques simples permettant une approche descriptive des phénomènes (calcul de la surface sous la courbe des concentrations par la méthode des trapèzes; pente d’élimination).
- Avec l’approche compartimentale, les variations de concentration sont décrites à l'aide de modèles mathématiques complexes permettant une description de phénomènes dynamiques. Dans ce cas, l’organisme est représenté par un ou plusieurs compartiments (figure 4) :
- Si l’organisme se comporte comme un ensemble homogène, la cinétique du médicament sera représentée par un modèle monocompartimental
- Si l’organisme se comporte comme plusieurs ensembles homogènes, la cinétique du médicament sera représentée par un modèle bi ou pluricompartimental avec un compartiment central et un ou plusieurs compartiments périphériques.

Figure 4: notion de compartiments. L’organisme est considéré comme une succession de compartiments dans lesquels le médicament se distribue et diffuse de l’un à l’autre.
3- Notion de linéarité de la pharmacocinétique
La pharmacocinétique d’un médicament est dite linéaire si les paramètres pharmacocinétiques ne varient pas avec la dose. C’est le cas de la majorité des médicaments.
- Dans le cadre d’une cinétique linéaire, l’évolution des concentrations du médicament est prévisible pour tout schéma posologique, à condition bien sûr qu’aucun événement intercurrent ne vienne modifier les paramètres individuels.
- Les médicaments dont la pharmacocinétique n’est pas linéaire sont difficiles à manier car les modifications de posologie s’accompagnent de variations imprévisibles des concentrations. La non-linéarité des cinétiques résulte le plus souvent de la saturation de l’un des processus pharmacocinétiques (ex. phénytoïne et saturation du métabolisme).
- Dernière mise à jour le .