
Biodisponibilité
L’administration d’un médicament par voie i .v. est réservée aux situations où un effet rapide est recherché ou aux médicaments qui ne peuvent pas être administrés par voie extravasculaire car peu ou mal absorbés. L’étape d’absorption existe pour toutes les voies d’administration extra-vasculaire (voie orale, cutanée, intra-musculaire, pulmonaire,…). Elle peut s’accompagner d’une perte en médicament, correspondant à une fraction non absorbée et qui n’atteindra pas la circulation générale. La phase d’absorption peut être limitante et l’étude de ce processus est indispensable et obligatoire pour chaque voie d’administration envisagée.
1. Cinétique après administration extravasculaire et biodisponibilité
Après administration d’une dose unique par voie extra-vasculaire, la concentration n’augmente pas instantanément comme dans le cas d’une administration i.v. car le médicament doit franchir des barrières biologiques avant d’arriver à la circulation générale (figure 1).
Figure 1: Evolution des concentrations sanguines du médicament après administration extravasculaire
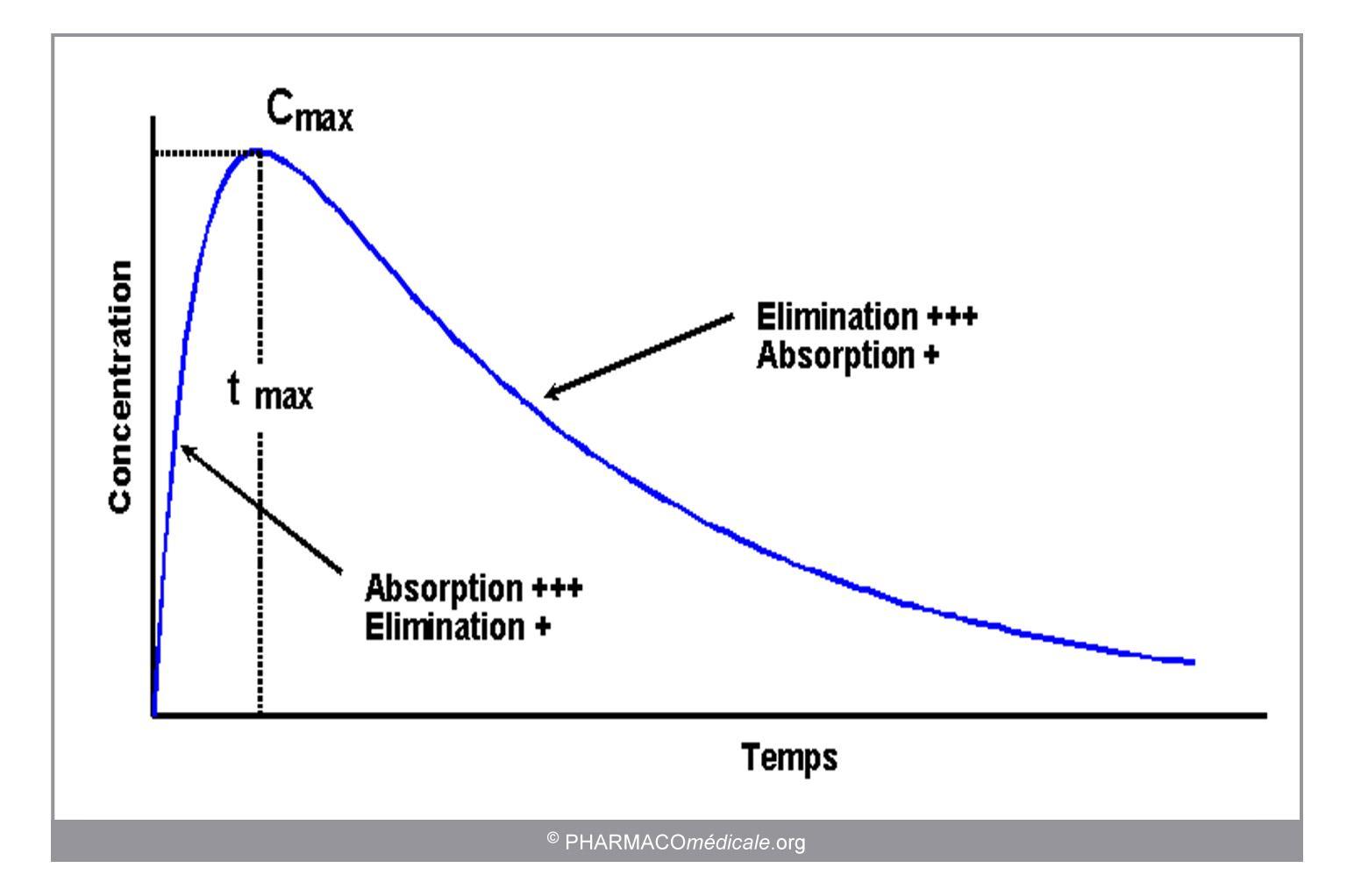
- L’évolution des concentrations au cours du temps est la résultante de l’entrée du médicament dans l’organisme et de son élimination. A la différence de l’administration i.v. unique, les processus d’absorption et d’élimination coexistent et l’aspect de la courbe variera avec les durées respectives de chacune de ces phases :
- Phase d’augmentation des concentrations : les concentrations augmentent tant que l’absorption est plus importante que l’élimination
- Pic de concentration : à un niveau de concentration donné, la vitesse d’élimination est égale à la vitesse d’absorption et la concentration atteint une valeur maximale (Cmax).
- Phase de décroissance des concentrations : les processus d’absorption et d’élimination coexistent toujours mais la vitesse d’élimination est supérieure à la vitesse d’absorption.
- L’évolution des concentrations au cours du temps répond à l’équation :
C = -A.e-kat + B.e-ket
La constante d’absorption ka inclut toutes les variations dues à la forme pharmaceutique et au passage transmembranaire. Ainsi, l’absorption d’un médicament dépend de :
- La voie d’administration ; un même médicament sera absorbé différemment par voie orale, rectale, cutanée…
- La forme galénique (comprimé, gélule, forme liquide), qui va modifier la cinétique de libération du principe actif et donc sa mise à disposition pour l’absorption.
- La biodisponibilité est définie par la quantité de médicament qui atteint la circulation sanguine après administration extravasculaire et par la vitesse de ce phénomène, qui dépend de la vitesse d’absorption à partir du site d’administration.
- La fraction biodisponible est exprimée par le facteur F. C’est un pourcentage pouvant varier de 0 à 100%.
- Le facteur vitesse est apprécié par la concentration maximale (Cmax) atteinte et le délai (Tmax) d’obtention de cette concentration maximale.
- La biodisponibilité d’un même principe actif peut donc être variable; elle est définie pour une voie d’administration et une formulation pharmaceutique données.
On distingue :
- La biodisponibilité absolue, correspondant au rapport de la quantité absorbée par une voie d’administration donnée à celle obtenue par voie i.v. (égale à 100%, par définition).
- La biodisponibilité relative, permettant de comparer entre elles deux formes du médicament administrées par la même voie (ex. comprimé vs sirop). La comparaison porte alors sur les 3 paramètres : F, Cmax et Tmax. La démonstration que ces paramètres sont comparables permet de définir la bioéquivalence des deux formes. La bioéquivalence des formes sera un critère essentiel pour la reconnaissance d’un médicament générique.
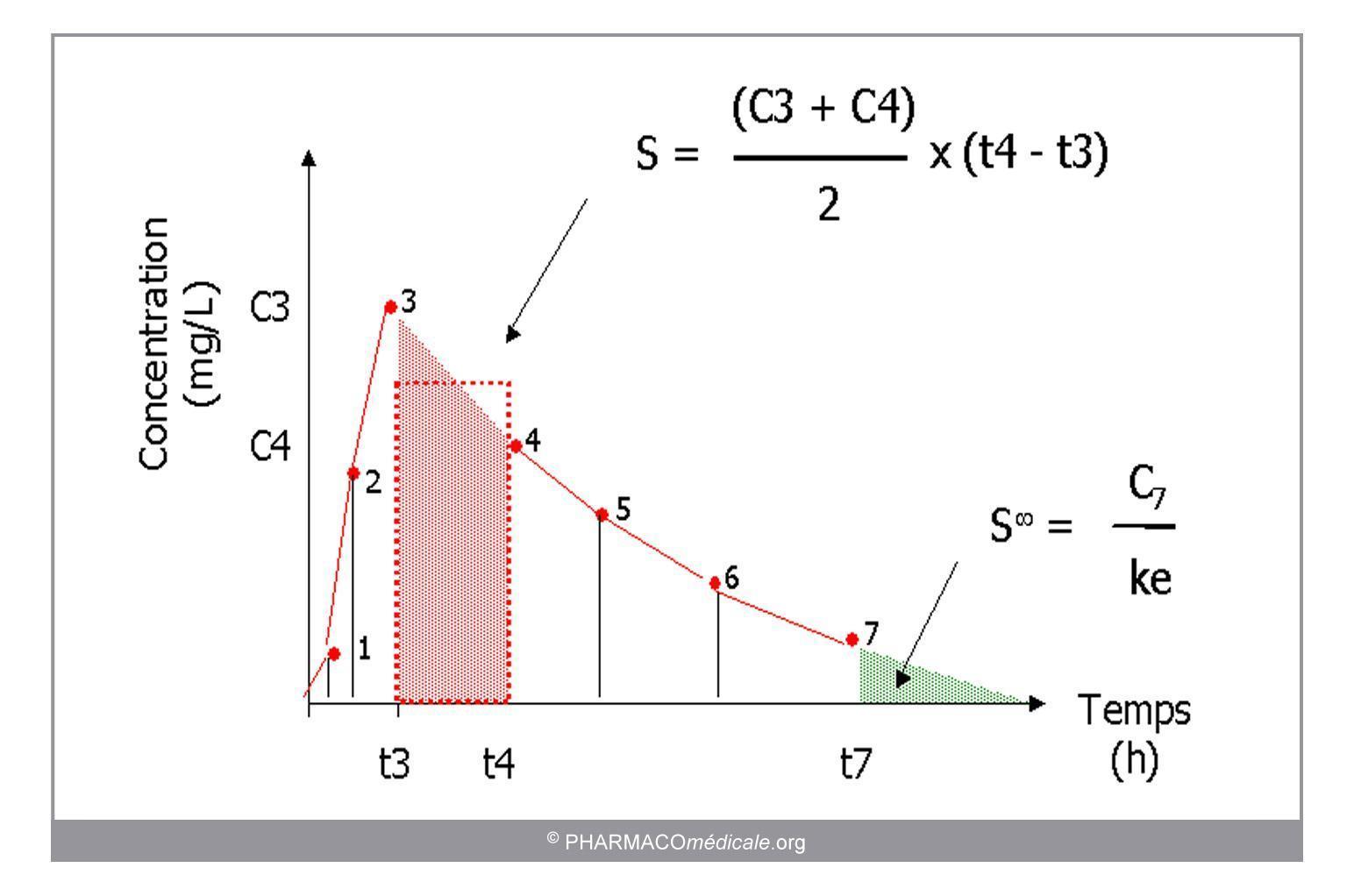
- La quantité de médicament ayant atteint la circulation générale peut être quantifiée par la surface sous la courbe (SSC), qui représente l’exposition de l’organisme au médicament.
La SSC peut être obtenue :
- Par résolution d’équation : SSC de 0 à l'infini = C0/ke en système monocompartimental i.v.
- Par analyse graphique, en utilisant la méthode des trapèzes (somme des trapèzes individuels). La SSC est exprimée en unité de concentration x unité de temps, par exemple : h.mg/L
FIgure 2 : Calcul de la surface sous la courbe (SSC) par la méthode des trapèzes
2. Mode de calcul de la biodisponibilité
- La biodisponibilité absolue d’un médicament est déterminée par référence à l’administration i .v. de ce même médicament pour laquelle, par définition, la biodisponibilité est totale et immédiate. On l’obtient par la relation :
F (%) = [SSCforme étudiée / SSCi.v.] x 100
Eventuellement corrigée du rapport des doses administrées si celles-ci sont différentes :
F(%) = [Dose i.v. x SSCforme étudiée / Dose forme étudiée x SSCi.v.] x 100
Figure 3 : Biodisponibilité par voie orale de 3 médicaments
- La biodisponibilité absolue sert à calculer le rapport de dose à administrer lors du relais de la voie i.v. par une autre voie d’administration. Par exemple, si la biodisponibilité orale du médicament est de 50%, on devra administrer pour cette voie une dose double de celle de la voie i.v.
- La biodisponibilité relative d’un médicament est déterminée par rapport à une autre forme pharmaceutique administrée par la même voie.
L’étude de biodisponibilité relative ou la recherche de bioéquivalence, comprend :
- La détermination de la fraction absorbée relative (FR), avec :
FR(%) = [Dose forme de référence x SSCforme étudiée / Dose forme étudiée x SSC forme de référence] x 100
- La comparaison des Cmax et des Tmax de chacune des formes étudiées.
L’écart maximum admissible sur chaque paramètre pour accepter la bioéquivalence de 2 médicaments est de 20%. Ces critères doivent impérativement (obligation réglementaire) être remplis pour la reconnaissance et l’enregistrement d’un médicament générique.
- Dernière mise à jour le .